





Abstract
The role of transcription in oligonucleotide (ODN)‐directed gene modification has been investigated in mammalian cells. The importance of transcription is demonstrated using mammalian cell lines with varying degrees of transcription of the mutant LacZ reporter gene, residing in both episome and chromosome. Gene correction occurs more efficiently when the target gene is actively transcribed and antisense ODN is more active than sense ODN. Using an approach that combines biochemical studies with a cell‐based assay to measure the functional activity of intermediates it is shown that a joint molecule, consisting of supercoiled DNA and homologous ODN targeted to correct the mutated base, is a functional intermediate in the gene repair process. Furthermore, this approach showed that a resected joint molecule is a downstream intermediate of the D‐loop. These results indicate that the primary reason for efficient gene repair exhibited by the antisense ODN is its increased accessibility to the non‐transcribed strand, and as a consequence an increased formation of intermediate during active transcription. Moreover, the processing of intermediates was also affected by transcription, suggesting that ODN‐directed gene repair may be linked to transcription‐coupled repair. Thus, transcription plays an important role in ODN‐directed gene repair by affecting the formation and processing of key intermediates.
Received January 8, 2003; Revised and Accepted February 21, 2003
INTRODUCTION
Gene alteration by oligonucleotides can be used for targeted alterations in the genome of eukaryotic cells. This approach has a potential to correct or introduce a point mutation in the targeted genomic DNA sequence while maintaining the genomic organization important for appropriate expression and regulation of gene. Several different strategies have been used to make a sequence‐specific gene alteration in mammalian cells, yeasts, plants and animals (see reviews 1–3 and references therein). These methods will become valuable in non‐viral gene therapy as well as in the identification of protein functions, once relatively low frequency of chromosomal alteration can be overcome. Besides the well known requirements, like the quality and delivery of oligonucleotides to cell, one needs to consider many other factors for successful gene repair. It is well known that the biological activities of cells, including DNA repair and recombination, and the status of replication and transcription of targeted genes play important roles. Moreover, each cell type has different repair activity and needs to be tested for its repair activity prior to extensive gene targeting experiments (4). On the other hand, given the low frequency of oligonucleotide‐directed gene alteration, a selection procedure is ultimately needed to make the gene repair technology practical.
Our laboratory has focused on two aspects of this research, the development of novel gene targeting approaches and mechanistic studies (4–8). Toward these goals, we have established the mutant LacZ (mtLacZ) system, which provides clear evidence of gene correction by oligonucleotides in the episome and chromosome of mammalian cells (7). Using a biochemical system where nuclear extracts from mammalian cells were capable of gene correction, we showed that there was a large variation in frequencies of gene correction among different cell types (4). Recently, we found that short single‐stranded oligodeoxynucleotides (ODNs) were capable of gene correction in CHO‐K1 cells (7), confirming previous findings of DNA alterations by ODN in mammalian cells (9,10) and yeast (11,12). Very recently, we showed that ODN was capable of correcting an integrated copy of the mtLacZ sequence in mouse embryonic stem cells (13). In another recent study, we showed that two ODNs designed to produce alterations in tyrosinase and c‐kit genes, respectively, can be used to cause simultaneous modifications of both genes in a single albino mouse melanocyte (14). Our results indicate that if two ODNs are present within the nucleus of a ‘repair‐competent’ cell, then dual targeting events can occur at relatively high frequency. Such a strategy may allow the use of selection procedure to overcome the low frequency of gene correction, a limitation of the current oligonucleotide‐based gene targeting.
During our investigation of ODN‐directed gene modification, we made the first observation of preferentially enhanced gene repair activity of antisense ODN over sense ODN and suggested a potential effect of transcription on gene repair by ODN (7). This result has been subsequently verified in yeast (15,16). Initially, we proposed that the preferential action of antisense ODN could arise from DNA strand separation during transcription and, as a consequence, its increased accessibility to the non‐transcribed strand. Another possibility was that the sense and antisense ODN had equal accessibility but antisense showed more efficient repair due to the strand specificity of the DNA repair system. The recent study by Liu et al. (16) reported that such strand bias in yeast was caused by displacement of the sense ODN by RNA polymerase during active transcription. It is not known if these findings apply to mammalian cells.
In this study, we report the role of transcription in ODN‐directed gene repair of the mtLacZsequence residing in the episome or chromosome of mammalian cells. Our findings indicate that gene correction occurs more efficiently when the target gene is actively transcribed and antisense ODN is more active than sense ODN. We also investigated the mechanism responsible for the enhanced activity of antisense ODN over sense ODN. A unique advantage of our system is that biochemical activity and functional activity can be addressed in the same system. We demonstrate that mammalian cells can process a joint molecule consisting of supercoiled DNA and homologous ODN resulting in correction of the mutated base. Furthermore, we show that a resected joint molecule is a downstream intermediate in the process. These mechanistic studies also lead us to conclude that the primary reason for the preferential action of the antisense ODN is increased accessibility of antisense ODN to the non‐transcribed strand caused by strand separation during transcription. Our results suggest that ODN‐directed gene repair may be linked to transcription‐coupled repair (TCR).
MATERIALS AND METHODS
Plasmids and oligonucleotides
The mammalian shuttle vectors pCH110‐G1651A, pRetro‐On‐mtLacZ and pTRE2Hyg‐mtLacZ contained the LacZ gene with an inactivating point mutation (G to A), which results in an amino acid substitution of E526K that inactivates the β‐galactosidase activity completely. The construction of the original pCH110‐G1651A plasmid has been described previously (4). The pRetro‐On‐mtLacZ and pTRE2Hyg‐mtLacZ plasmids were generated by inserting a 3.7 kb fragment containing the full‐length of the mtLacZ gene from the pCH110‐G1651A plasmid into NotI and BamHI sites of the pRetro‐On plasmid (Clontech, Palo Alto, CA) and the pTRE2Hyg plasmid (Clontech), respectively. Plasmid DNAs were prepared by large‐scale DNA preparation (Qiagen, Santa Clarita, CA) and extensively dialyzed before use.
The oligonucleotides were synthesized by the standard phosphoramidite procedure using a 1 µmol scale (Nucleic Acid Core Facility, University of Pennsylvania, PA) and purified by HPLC.
Cells and tissue culture conditions
F12K medium and fetal bovine serum (FBS) were purchased from Cellgro (Herndon, VA) and Biowhittaker (Wakersville, MD), respectively. Alpha‐MEM medium was from Cellgro. All media were supplemented with 2 mM l‐glutamine. CHO‐K1 cells (ATCC, Rockville, MD) were maintained in F12K medium containing 10% heat‐inactivated FBS. CHO‐AA8‐Luc cells (Clontech) were grown in Alpha‐MEM medium supplemented with 10% FBS. CHO‐K1‐mtLacZ cells were generated in our laboratory as described previously (7) and maintained in F12K medium containing 10% FBS. All tissue culture cells were grown at 37°C and 5% CO2.
Transfection and detection of gene correction
For the episomal targeting, 0.5 × 105 cells (CHO‐K1 or CHO‐AA8‐Luc) were seeded per well in a 6‐well plate 16–18 h before transfection. Oligonucleotide (4.8 µg) and plasmid DNA (2.0 µg) were pre‐incubated with Lipofectamine (Gibco, Bethesda, MD) at a ratio of 1:1 (w/w) in 200 µl of OptiMEM for 45 min at room temperature. The DNA/liposome complex was added to cells in a final volume of 1.0 ml, made up with OptiMEM. Six hours later, cells were fed with 2.0 ml of the complete growth medium and stained 48 h after transfection. Where required, 0.5 µg/ml of doxycycline (Dox) was added to cells. Because Dox has a relatively short half‐life time, it was replaced every 8–16 h. Gene correction was detected by histochemical staining of cells. Prior to the staining, cells were washed two times with cold PBS and fixed for 5 min in 1% glutaraldehyde at 4°C. After removal of fixation solution, cells were extensively washed with PBS and then stained with the X‐gal solution [5.0 mM K3F3(CN)6, 5.0 mM K4F2(CN)6, 2.0 mM MgCl2 and 1.0 mM 5‐bromo‐4‐chloro‐3‐indolyl‐β‐d‐galactoside (X‐Gal)] in PBS (pH 7.4) at 37°C overnight. Blue cells were counted for each well using light microscopy. To count as many as 2500 cells per one well of a 6‐well plate, we divided one well into four sections and counted each blue cell at 10× magnification. Frequency of correction was measured by dividing the number of positively stained cells by the initial number of cells plated.
For the chromosomal targeting, 0.5 × 105 CHO‐K1‐mtLacZ cells were seeded in a 6‐well plate, 16–18 h before transfection. Oligonucleotide (7.5 µg) was diluted to 100 µl with OptiMEM and pre‐incubated with 7.5 µg of Lipofectamine in a final volume of 200 µl. The complex was allowed to form for 45 min and added to cells in a final volume of 1.0 ml, made up with OptiMEM. Cells were fed with 2.0 ml of complete medium 6 h later and stained with X‐Gal solution 48 h after transfection as described above.
Luciferase assay
For detection of luciferase activity in the CHO‐AA8‐Luc cell line, cells were transfected with pTRE2Hyg‐mtLacZ (2.0 µg) and/or oligonucleotide (4.8 µg) as described above. After the removal of DNA/liposome complex, cells were fed with 2.0 ml growth medium with or without 0.5 µg/ml Dox and the luciferase assay was carried out 48 h after transfection. During incubation, Dox was replaced every 16 h. Luciferase activity was measured according to the manufacturer’s specification (Clontech). Briefly, cells were washed twice with 1× PBS and lysed in a 200 µl of 1× lysis buffer by shaking at room temperature for 15–20 min. Cellular debris was removed via centrifugation and 50 µl of the final lysate was assayed using the Luminometer (EG & G Berthold Systems, Aliquippa, PA).
Hirt DNA analysis
General procedures of episomal DNA (Hirt) isolation and bacterial read‐out assays were described previously (4). Briefly, the rescued Hirt DNA from the transfected cells was electroporated into the P90C Escherichia coli strain. Transformation of the plasmid containing corrected or mutant LacZ gene results in either blue or white colonies on Amp/X‐Gal plate, respectively. Frequency of correction was measured by dividing the total number of blue colonies by the total number colonies growing on the X‐Gal/Amp plate. To verify gene correction at the DNA sequence level, plasmid DNAs from blue and white colonies were isolated and subjected to the PCR–RFLP analysis, as described previously (4). Then, purified PCR products were subjected to direct DNA sequencing by automatic DNA sequencer (ABI 373 A, Applied Biosystems).
Southern blot analysis
CHO‐K1‐mtLacZ cells (clones 8 and 14) were grown until confluent and then lysed with a buffer containing 100 mM Tris–HCl, pH 8.5, 5 mM EDTA, 200 mM NaCl, 0.2% SDS and 100 µg/ml proteinase K at 56°C overnight. Genomic DNA was purified twice with phenol/chloroform and once by chloroform extraction followed by isopropanol precipitation. Ten micrograms of the genomic DNA from clones 8 and 14 were subjected to double digestion by restriction enzymes PvuII and HindIII, which generated a 2.5 kb fragment, residing within the coding region of β‐galactosidase. To measure the copy number of integrated mtLacZ, the same genomic DNA was digested with HindIII, which has only one site within the coding region of β‐galactosidase. Southern analysis was performed by a standard procedure. Digested DNA was electrophoresed through 0.7% agarose gel with subsequent depurinization, denaturation and neutralization. Then, DNA was transferred onto Zeta Probe nitrocellulose membrane (Bio‐Rad, Hercules, CA), cross‐linked and probed with the 32P‐labeled 2.5 kb DNA fragment (specific activity of 1 × 106 c.p.m./ml in the hybridization buffer) isolated from the pCH110‐G1651A plasmid. The hybridization signal was detected by autoradiography at –80°C.
Northern blot analysis
CHO‐K1‐mtLacZ cells (clones 8 and 14) were grown until confluent and then total RNA was extracted using the Qiagen RNAesy extraction kit (Valencia, CA). Twenty micrograms of total RNA was electrophoresed through 1.0% denaturing formaldehyde agarose gel in 1× MOPS buffer (Ambion, Austin, TX). The gel was denatured in 50 mM NaOH solution for 10 min and then neutralized in 10× SSC buffer. Then, RNA was transferred to Zeta Probe membrane, cross‐linked, and probed with the 32P‐labeled 2.5 kb DNA fragment (specific activity of 1 × 106 c.p.m./ml). Hybridization signal was detected by autoradiography at –80°C.
Western blot analysis
Cells were lysed in a buffer containing 50 mM Tris–HCl at pH 7.4, 1% Na‐deoxycholate, 1% Triton X‐100, 0.1% SDS, 0.15 mM NaCl, 1.5 mM EDTA and protease inhibitors. Proteins were separated by 7% SDS–PAGE and transferred to nitrocellulose membrane followed by the western blot analysis using a monoclonal anti‐β‐galactosidase antibody (Promega, Madison, WI) and anti‐mouse secondary antibodies labeled with the alkaline phosphatase (Promega).
D‐loop formation by RecA and its functional assay
The formation of synaptic complexes was carried out between RecA‐ODN filament and the duplex plasmid DNA, pCH110‐G1651A or pTRE2Hyg‐mtLacZ, respectively. Initially, the 32P 5′‐labeled ODN (84 nM) was pre‐incubated with 2.0 µg of E.coli RecA protein (New England Biolabs, Beverly, MA) in a reaction buffer containing Tris–HCl at pH 7.4, 1 mM DTT, 1.0 mM Mg(OAc)2, 0.3 mM ATPγS, 100 mg/ml BSA in a total volume of 20 µl for 15 min at 37°C. All oligonucleotides used were 32P 5′‐end‐labeled by T4 kinase with a specific activity of 1017 c.p.m./mol. After the pre‐synaptic filament formation, the strand exchange reaction was performed with pCH110‐G1651A or pTRE2Hyg‐mtLacZ plasmid (21 nM), respectively, in 10 mM Mg(OAc)2 for 5 min at 37°C. Synaptic complexes were deproteinized by addition of 1% SDS for 15 min and an entire sample was analyzed by 0.8% agarose gel electrophoresis in TBE buffer containing 2 mM MgCl2 at 4°C. The gel was visualized by ethidium‐bromide staining and subsequently dried. Joint molecules were detected by autoradiography.
For transfection of the preformed joint, the D‐loop formation was performed using a 4:1 molar ratio of the preformed RecA‐ODN filament to the target plasmid DNA, either pCH110‐G1651A or pTRE2Hyg‐mtLacZ. Synaptic complexes were deproteinized by addition of 1% SDS for 15 min. RecA and SDS were subsequently removed from the reaction mixture by precipitation with 100 mM KCl. The joint complexes were further purified from unbound ODN using the Qiagen PCR purification kit (Qiagen, Santa Clarita, CA). The presence of D‐loop was confirmed by agarose gel electrophoresis and autoradiography as described above.
Equal amounts of pCH110‐G1651A/ODN and pTRE2Hyg‐mtLacZ/ODN joints, respectively, were used to transfect CHO‐K1 or CHO‐AA8‐Luc cells. Gene correction was detected by histochemical staining with the X‐Gal solution 48 h after transfection of joint molecules by scoring cells with positive β‐galactosidase activity.
S1 nuclease cleavage of D‐loop and functional assay
Initial formation of D‐loop and purification were carried out as described above. Purified joint molecules consisting of pCH110‐G1651A plasmid DNA and X1 or X2 ODN were incubated at various concentrations of S1 nuclease (0.1, 1 and 10 U/µl) for 30 min at 37°C. S1 nuclease at 0.1 U/µl converted 80% of DNA from supercoiled to relaxed circle DNA without further degradation as detected by ethidium‐bromide staining. The reaction was stopped by the addition of 25 mM EDTA. Resected D‐loop was then purified using CHROMA SPIN‐400 column (Clontech) and assayed on 0.8% agarose gel followed by ethidium‐bromide staining. Equal amounts of resected joints were transfected into CHO‐K1 cells. Gene correction was detected by histochemical staining with X‐Gal solution.
RESULTS
Episomal gene correction by ODN is dependent on transcription
We have found that ODN in antisense orientation showed a preferentially enhanced gene repair activity than ODN in sense orientation in mammalian cells and suggested a potential effect of transcription on gene repair (7). To further investigate the effect of transcription in gene correction by ODN, we constructed two plasmids in which transcription of mtLacZ can be regulated, using the Tet‐On and Tet‐Off systems (Fig. 1A). Sequences of synthetic oligonucleotides used in this study are shown in Figure 1B.
Tet‐On
In the Tet‐On system, we used pRetro‐On‐mtLacZ plasmid, which encodes both mtLacZ gene under the control of tetracycline response element (TRE) and reverse transcriptional activator (rtTA) under the control of SV40 promoter within the same plasmid. The addition of Dox to culture medium of cells transfected with pRetro‐On‐mtLacZ plasmid induces binding of rtTA to the TRE sequence and results in transcriptional activation of mtLacZ. To investigate whether the extent of induction was dependent on the amount of transfected plasmid, different amounts (2–12 µg) of pRetro‐On‐mtLacZ plasmid were used in each transfection experiment. At a higher concentration of plasmid (6 and 12 µg), the extent of induction was not as pronounced as that observed using 2 µg of plasmid, detected by the western blot (data not shown). The molar amount of transfected plasmid is likely to exceed available Tet activators present in cells at high concentrations. Based on this result, all experiments were carried out with 2 µg of plasmid.
For the episomal targeting, transient transfection of pRetro‐On‐mtLacZ plasmid and X1 (antisense) or X2 (sense) ODN was performed in CHO‐K1 cells. In all experiments, transfections were carried out in duplicate where each set of transfected cells was either treated with Dox or untreated. As shown in Table 1, both X1 and X2 ODN showed a 10‐fold increase in gene correction when transcription of mtLacZ was induced. Furthermore, X1 ODN was 2‐fold more active than X2, regardless of the level of transcription.
Tet‐Off
To verify our findings using a converse approach, we constructed pTRE2Hyg‐mtLacZ plasmid for the Tet‐Off system. In this case, the Tet‐responsive transcriptional activator (tTA) binds to the TRE in the absence of Dox and activates transcription. Thus, addition of Dox to the culture medium turns off transcription of mtLacZ.
For transfection, we utilized the CHO‐AA8‐Luc cell line containing an integrated luciferase gene under TRE control. Expression of luciferase was shown to be tightly regulated by the tTA present in these cells (10 000‐fold), according to the manufacturer’s specification. Therefore, the level of luciferase expression was measured in all our experiments and served as an internal control for the extent of transcriptional regulation. Initially, several concentrations of Dox were examined to down‐regulate luciferase expression in CHO‐AA8‐Luc cells (data not shown). The most potent concentration (0.5 µg/ml) was used in all further experiments. To ensure that the presence of transfected pTRE2Hyg‐mtLacZ plasmid and ODN in these cells did not affect the endogenous luciferase gene expression, which is also under the control of TRE, we measured luciferase activity for each transfection experiment and showed that luciferase activity was sufficiently inhibited ∼300–500 times with Dox in all our experiments (data not shown).
Similar to data obtained in the Tet‐On system, the cells transfected without Dox showed a high level of gene conversion (Table 1). Once again, we found a reduction in gene correction when the transcription of mtLacZ was down‐regulated. Furthermore, X1 ODN was more active than X2 ODN, similar to the Tet‐On system. The low level of correction, which is seen in transfections with down‐regulated transcription for both systems, was caused by residual transcription that could not be completely inhibited. This residual transcription was sufficient to cause different gene repair activity of sense and antisense ODNs in both systems.
To verify gene correction at the DNA sequence level, Hirt DNA was isolated from the transiently transfected cells and analyzed as previously described (4). Both pRetro‐On‐mtLacZ and pTRE2Hyg‐mtLacZ plasmids contain both eukaryotic (SV40) and bacterial guanosine phosphoryl transferase (gpt) promoters for mtLacZ expression. The Hirt DNA was recovered after transfection and electroporated into the E.coli strain P90C. The ratios of blue to white bacterial colonies were 10–5 for both plasmids which were at least 1000‐fold lower than those found in mammalian cells. This difference indicates that mammalian cells can take up a large number of plasmids per cell and can be scored as positive blue cells even when a single plasmid per cell has been corrected. In a given system, nevertheless, the histochemical staining of mammalian cells can be used to compare the relative activities of sense and antisense ODNs. Sequence analysis of blue bacterial colonies generated by Hirt DNA confirmed the correction of AAA to GAA at position 1651 and no other sequence change was observed in a 300 bp region flanking the mutated site (data not shown). Thus, X1 and X2 ODN corrected the mtLacZ gene in a site‐specific manner.
Chromosomal gene correction by ODN is dependent on transcription
To further investigate the role of transcription in the gene conversion process of chromosomal DNA, we generated two stable CHO‐K1 cell lines (CHO‐K1‐mtLacZ) with varying degrees of transcription of the integrated mtLacZ gene. The presence of transgene in these clones was verified by PCR amplification of genomic DNA and the copy number of integrated mtLacZ was determined by Southern blot analysis (Fig. 2A). To demonstrate the consequences of transgene integration at the mRNA and protein level, clones were analyzed by northern and western blot analyses (Fig. 2B and C). As can be seen, each clone contained a single copy of the integrated mtLacZ but both mRNA and protein levels were reduced 20‐fold, respectively, in clone 14 in comparison to clone 8.
In order to examine whether the extent of gene correction is related to the level of transcription of mtLacZ, chromosomal targeting was performed using either antisense (X1) or sense (X2) ODN with a 45 nt homology to the target chromosomal sequence. Results obtained from five separate targeting experiments are summarized in Table 2. The X1 ODN showed a 100‐fold higher gene correction frequency in clone 8, when compared to clone 14. Both clones showed similar transfection efficiency when tested by transfection of wild‐type LacZ plasmid or fluorescent‐conjugated ODN (data not shown). Thus, we conclude that the decrease in the rate of gene repair in clone 14 was caused by a low level of transcription of mtLacZ. When X2 ODN was introduced, similar results were obtained; the frequency of correction was at least 30‐fold higher in clone 8 than in clone 14. Once again, we confirmed that antisense ODN showed ∼10‐fold higher gene correction activity than sense ODN in cells with high level of transcription (clone 8). However, even antisense ODN failed to show any significant level of chromosomal gene correction in cells with low transcriptional activity (clone 14, see Table 2). According to our data, we conclude that antisense ODN facilitates efficient gene repair of a point mutation in highly transcribed genes. In addition, our results also suggest that antisense ODN may not facilitate efficient repair in genes with repressed transcription.
Formation of the D‐loop by RecA
It is hypothesized that the crucial reaction for gene correction by ODN involves a strand pairing of ODN to the homologous DNA, leading to formation of a D‐loop. Our previous study with nuclear extracts indicated that the formation and the processing of joint molecules (D‐loop) are important factors regulating gene repair (8). Based on these results, we tested whether the enhanced gene repair during transcription might be caused by an increase in the amount of joint molecule formation facilitated by strand invasion of ODN. However, this assumption will be valid only if joint molecules are functional intermediates in the gene repair process. Therefore, we first preformed joints by using RecA protein to facilitate formation of synaptic complex and then transfected a known amount of joints directly into mammalian cells to score gene correction.
A joint molecule was made between the supercoiled pCH110G1056A plasmid DNA and the antisense (X1) or sense ODN (X2). Optimization of the conditions for pre‐synaptic filament formation was done by varying the amount of RecA protein (1.0, 2.0 and 4.0 µg) and the incubation time (5, 15 and 30 min) (data not shown). Based on these results, 32P 5′‐labeled ODN (84 nM) was first incubated with 2.0 µg of RecA protein for 15 min. Next, 21 nM of a homologous plasmid DNA (pCH110G1056A) was added to this pre‐synaptic filament and further incubated for 5 min. The ability of RecA protein to catalyze the incorporation of the 32P 5′‐labeled ODN into homologous pCH110‐G1651A plasmid was monitored by agarose gel electrophoresis after RecA removal followed by autoradiography. Absence of any incorporated radioactivity seen in Figure 3A indicates that D‐loop formation is completely dependent on the presence of RecA. Moreover, the X1 and X2 ODNs made equal amounts of joints with the supercoiled DNA (Fig. 3B). The more slowly migrating DNA species represent either a nicked or a relaxed circle. These species also promoted the ODN assimilation, although at a reduced efficiency (20%) relative to the supercoiled DNA. The efficiency of the complex formation by RecA was ∼25–35% as estimated by counting the radioactivity incorporated into the supercoiled DNA.
Since our primary task was to isolate joint complexes for transfection in mammalian cells, the unbound ODN was removed from the joints as described in the Materials and Methods. Quantitation showed that ∼70% of the initial joints survived the purification procedure and were suitable for further experimentation (Fig. 3C). In addition, we have determined the stability of D‐loop complex. The D‐loop was stable at 37°C (Fig. 3D). Thus, the transfected D‐loop will survive inside the cell.
D‐loop is a functional intermediate
To test the functional activity of isolated D‐loop intermediates, equal amounts of purified joints (estimated by the radioactivity associated with supercoiled DNA) were introduced into CHO‐K1 cells (Fig. 4). The X1‐JM or X2‐JM designates the purified D‐loop complex between the pCH110‐G1651A plasmid and X1 or X2 ODN, respectively. Surprisingly, transfection of the preformed X2‐JM showed two times higher gene repair activity than X1‐JM, contrary to the results obtained from the cotransfection experiment (Fig. 4A). As a control, the plasmid and ODN were separately introduced to CHO‐K1 cells by conventional cotransfection. As expected, co‐transfected X1 ODN showed seven times higher activity than X2 ODN (Fig. 4A), consistent with our previous data (7). Meaningful comparisons of gene correction can be made between cotransfection and JM transfection experiments, because we used the same amount of plasmid in both cases. The opposite results obtained from cotransfection and JM transfection indicate that their difference is caused by the presence of transfected joint molecules in living cells.
When equal amounts of X1‐JM and X2‐JM, respectively, were introduced into CHO‐AA8‐Luc cells, X2‐JM again showed 12 times higher gene repair activity than X1‐JM (Fig. 4B). To further support this observation, the preformed joint molecules were tested in the Tet‐Off system. The joints were formed between ODN and pTRE2hyg‐mtLacZ plasmid and then transfected into CHO‐AA8‐Luc cells in the presence or absence of Dox. Once again, the X2‐JM showed five times higher activity than X1‐JM, when transcription was induced (Fig. 4C). The consistent findings observed in these three different examples strongly suggest that X2‐JM, in which sense ODN is hybridized to the transcribed strand, is processed more efficiently for repair than X1‐JM in living cells, indicating a preferential processing of X2‐JM by selective repair of the transcribed strand. Furthermore, it indicates that the primary reason for the higher repair rate observed with antisense ODN in cotransfection is its increased accessibility to the target DNA.
Resected D‐loop is a downstream functional intermediate
The data above confirm that D‐loop is a functional intermediate in oligonucleotide‐directed gene repair in mammalian cells. A cleavage in the single‐stranded region of D‐loop was shown to stabilize the intermediate and advance the reaction toward a recombinant product (17). To investigate whether such a processed D‐loop could also be a functional intermediate, we first prepared the resected joint by making the D‐loop with RecA, isolating D‐loop from unbound ODN, and then treating D‐loop with S1 nuclease. The extent of resected D‐loop was analyzed by agarose gel electrophoresis (Fig. 5A). When S1 nuclease was added, ∼80% of the supercoiled DNA was converted to the more slowly migrating species, nicked circle. We then transfected equal amounts of the resected X1‐JM and X2‐JM, respectively, into CHO‐K1 cells and scored gene repair. Once again, the resected X2‐JM showed 17 times higher activity than the resected X1‐JM. As seen in Figure 5B, the resected X2‐JM showed a 9.5‐fold increase in gene repair when compared to the authentic D‐loop, while the resected X1‐JM did not produce an appreciable increase from the corresponding D‐loop. These results suggest that the resected D‐loop is an important functional intermediate downstream of the D‐loop and transcription also affects the processing of this downstream intermediate in a similar strand‐specific manner.
DISCUSSION
We demonstrated previously that ODN caused a sequence‐specific, homology length‐dependent and strand‐specific gene correction in mammalian cells (7). The optimal ODN length of 45 nt suggested that factors, such as the size and structure of transiently open chromatin, might play a role in the initiation of the repair process. Moreover, two ODNs with the same length but opposite polarity showed significant differences in gene correction frequency that revealed for the first time the potential effect of transcription on gene repair by ODN. It is conceivable that chromatin structure of the highly transcribed regions may be more accessible to the pairing event mediated by ODN than the regions with inactive transcription.
Here, we examined the role of transcription in ODN‐directed gene repair of the mutant reporter LacZ gene residing either in episome or chromosome of mammalian cells. Using transient transfection of a plasmid, in which its transcription can be regulated by tetracycline, we show that episomal gene correction occurs more efficiently when the target gene is actively transcribed and antisense ODN is more active than sense regardless of the transcription level of the target gene. We then investigated ODN‐directed gene correction of genomic DNA by comparing two clones with different transcriptional levels. Our data indicate: (i) gene repair by ODN occurs much more efficiently when the target gene is actively transcribed; (ii) antisense ODN is more active than sense when the gene is actively transcribed; (iii) a point mutation in the transcriptionally repressed genes cannot be efficiently repaired by ODN. These results imply that one needs to select actively transcribing genes for ODN‐directed gene targeting.
The correction frequency of genomic DNA was much lower than that of episome DNA, as we previously reported (7). However, the effect of transcription in gene correction was much more pronounced in targeting of genomic DNA than episome. Furthermore, the difference in correction rate by antisense and sense ODN was more dramatic in genomic DNA targeting than episome. These differences can be explained in part by leakiness of plasmid transcription in both the Tet‐On and Tet‐Off systems. In order to regulate transcription of genomic sequence more tightly, we compared two clones with a single copy of the integrated gene but very different levels of transcription. Although it is possible that different repair efficiency found in these clones could reflect locus‐specific effects or clonal variation of genomic DNA, consistent results found in episomal and chromosomal targeting suggest a general role of transcription in gene correction.
We then investigated the mechanism linking gene repair and transcription by combining a biochemical approach with a cell‐based assay to measure the functional activity of intermediates. The initial reaction for gene correction is hypothesized to involve strand invasion of ODN into the homologous DNA to form a D‐loop. Although such an intermediate was shown to be functional in E.coli (18), the presence of such an intermediate or its functional activity has not been demonstrated in mammalian cells. We recently showed that mammalian nuclear extracts promoted strand pairing of supercoiled DNA and its homologous ODN as an initial step (8). During these prior investigations, we observed that joint molecules were too stable to be an authentic D‐loop. While the D‐loop complex made by RecA dissociated completely at 65°C, 50% of joints made by nuclear extracts still remained at 65°C. This surprisingly high stability of the joint formed by nuclear extracts indicated that ODN formed an initial D‐loop but was subsequently hybridized to the complementary sequence in the plasmid via resection of the D‐loop by nucleases present in extracts (8).
Based on these results, we tested whether the D‐loop and resected D‐loop can be functional intermediates in the gene repair process by first preforming the joints and then scoring the gene repair rate of transfected joints in mammalian cells. Our data indicate that mammalian cells can process the preformed D‐loop and can correct the point mutation in the D‐loop. Moreover, we found that the resected D‐loop exhibited much higher gene repair activity than the D‐loop itself. These results suggest that the initially formed D‐loop could be further processed to a resected D‐loop, a more stable downstream intermediate for gene repair, consistent with our in vitro data (8).
To understand the molecular mechanism of the increased gene repair during active transcription, we have utilized the mtLacZ system where the functional gene repair activity of biochemical intermediates can be measured. To explain the preferential repair by antisense ODN, we considered three possibilities: (i) the opening of chromatin during transcription increases accessibility of the antisense ODN to the non‐transcribed strand preferentially; (ii) both the antisense and sense ODN have an equal accessibility to the target DNA, but the antisense ODN exhibits more efficient gene repair rate, because DNA repair systems operate in a strand‐specific manner; (iii) both the antisense and sense ODN have an equal accessibility to the target DNA but the sense ODN is preferentially displaced during transcription, thus leading to a low gene repair activity.
To distinguish between these possibilities, we first tested whether antisense ODN formed more joints than sense ODN during transcription and whether such an increase could cause enhanced gene repair activity of antisense ODN. During the mRNA chain elongation, an upstream portion of the non‐transcribed strand of ∼18 nt has been shown to lie on the outside of RNA polymerase complex with the bases exposed (19). This melted region of the non‐transcribed DNA forms a transcriptional bubble that could be accessible for preferential strand pairing by antisense ODN (Fig. 6, step 1). To test this possibility, equal amounts of preformed D‐loops (X1‐JM and X2‐JM) were transfected into mammalian cells to eliminate the difference caused by joint formation during transcription. Surprisingly, the preformed X2‐JM showed greater gene repair activity than X1‐JM. Likewise, the resected X2‐JM showed greater activity than resected X1‐JM. These results are opposite to our previous findings when the plasmid and ODN were introduced separately.
This surprising finding indicates that transcription plays a role in both formation and processing of intermediates. When equal amounts of joints were introduced, the joint made by sense ODN exhibits a higher correction rate than the joint by antisense ODN. Preferential repair by X2‐JM, in which sense ODN is hybridized to the transcribed strand, indicates that ODN‐directed gene repair may be linked to TCR that removes DNA lesions in the transcribed strand faster than those in the non‐transcribed strand (see reviews 20–22 and references therein). Thus, we conclude that the higher repair rate by antisense ODN is caused by its increased accessibility to the non‐transcribed strand during transcription and, as a consequence, an increase in the amount of D‐loop formation.
Our results clearly rule out the conclusion made by Liu et al. that lower correction rate by the sense ODN was caused by displacement of sense ODN by RNA polymerase (16). If this were the case, then X1‐JM should still exhibit a higher activity than X2‐JM. Their conclusion was based on the in vitro transcription of a joint molecule by the T7 polymerase, which may not bear resemblance to the cellular environment where the transcriptional strand bias of gene repair occurs (16). Our study clearly shows the importance of carrying out mechanistic studies in the same system where the strand specificity is observed, not in an artificial system.
The central role in the recruitment of DNA repair systems to the transcribed strand has been attributed to the transcription repair coupling factors, such as Cockayne Syndrome A (CSA) and B (CSB), yeast homolog Rad26 and bacterial protein Mfd, a family of ATPases with chromatin remodeling activities (23). These coupling factors displace the stalled RNA polymerase transiently and recruit proteins involved in DNA repair near the site of DNA damage (20,24). Multiple DNA repair systems may be coupled to transcription, including nucleotide excision repair (NER), base excision repair (BER) and mismatch repair (MMR). The mechanism of transcriptional coupled‐NER has been well established by sharing of common helicases, XPB and XPD, required for both NER and transcription (20). Recently, other DNA repair pathways, BER and MMR, have also shown to be coupled to transcription (21,25,26). For example, 8‐oxoguanine, which is usually repaired by BER, was rapidly removed from the transcribed strand but not the non‐transcribed strand, indicating that BER is also coupled with transcription (25). Moreover, unrepaired 8‐oxoguanine blocked transcription by RNA polymerase II and showed a high rate of mutations in fibroblasts isolated from CSB cells in comparison to normal fibroblasts. In addition, mismatch repair also appears to be linked to transcription. Several cell lines lacking mismatch repair genes, MSH2, PMS2 and MLH1, were found to be deficient in TCR in mammalian cells (26). Some features of the stalled RNA polymerase at the lesion and nascent RNA chain might resemble a mismatched heteroduplex to which mismatch repair proteins would bind. This could facilitate recognition and repair of the mismatched base in the transcribed strand selectively (21). What is clearly emerging is that transcription plays an important role in many different types of DNA repair pathways. Further studies are in progress to understand the role of transcriptional coupling factors and other transcription‐coupled DNA repair pathways in ODN‐directed gene repair.
Based on our results, we propose the following hypothetical model for ODN‐directed gene repair (Fig. 6). Step 1: during transcriptional elongation, an upstream portion of the non‐transcribed strand forms a transcription bubble that is accessible for strand pairing by the antisense ODN. In contrast, the transcribed strand is occupied by RNA polymerase complex, limiting accessibility of sense ODN to the transcribed strand. This step can be facilitated by the Rad52 epistasis group proteins including Rad51 (27–29). Recently, we showed that HsRad51 activity is necessary for the initial strand pairing in nuclear extracts from human cells (8). Steps 2 and 3: once the D‐loop is formed, trafficking of RNA polymerase is interrupted and the presence of stalled RNA polymerase near the D‐loop can signal for recruitment of DNA repair systems. The D‐loop is then converted to the resected D‐loop by nucleases (shown as arrows). One potential candidate for this nuclease is XPG protein, a structure‐specific 3′ endonuclease participating in NER (30), but also shown to play a distinctly different role in several other transcription‐coupled DNA repair pathways (21,25). Step 4: the presence of stalled RNA polymerase may serve as a signal to direct repair to the transcribed strand. In this case, the mismatch in the transcribed strand (T) formed by the sense ODN is preferentially repaired to C, leading to gene repair (A:T→G/T→ G:C). According to this model, the mismatch in the resected joint by antisense ODN (A/C) is more likely to be processed to A:T rather than G:C, producing no apparent gene repair. However, the observed gene correction can be explained in part by a novel short‐patch mismatch repair that efficiently corrects the A/C mismatch to G:C (31). This pathway is specific for the A/C mismatch only and differs from the conventional long‐patch mismatch repair (32). Although the joint made by sense ODN is processed to result in a higher gene repair activity than the joint by antisense, the most important factor is that antisense ODN formed much more joints to begin with than sense (see step 1 in Fig. 6). Thus, antisense ODN exhibits overall higher gene repair activity than sense, in spite of this directional processing. Moreover, the enhanced activity of antisense ODN holds for other mismatches, insertion and deletion (15), indicating a general transcription‐driven mechanism that operates regardless of the type of mismatch. However, it remains to be seen whether TCR can process intermediates by itself or requires additional DNA repair systems.
DNA replication has recently been implicated to cause a strand bias in homologous recombination of ODN in E.coli (33). The increased recombination efficiency of the ‘lagging strand’ ODN suggests that ODN recombination may occur near the replication fork and reflects more available single‐stranded regions during lagging versus leading strand synthesis. Because the plasmid DNA we used in this study does not replicate in mammalian cells, we can rule out the influence of replication in our episome targeting. However, it is possible that larger strand bias seen during chromosomal gene correction could in part be influenced by replication.
In conclusion, we show that transcription facilitates the formation of intermediates in the ODN‐directed gene repair process and also directs the recognition and repair of the mismatched base in the transcribed strand selectively. Gene correction occurs more efficiently when the target gene is actively transcribed. Thus, mechanistic studies to elucidate the link between transcription and ODN‐directed gene repair will have an important implication in the selection of genes to be targeted by ODN. Although the mechanism of gene repair by ODN in mammalian cells is not yet fully understood, our study represents the first step toward understand the role of transcriptional coupling factors and to explore other transcription‐coupled DNA repair pathways operating in ODN‐directed gene repair.
ACKNOWLEDGEMENTS
We thank Drs J. F. Klement and E. Pierce for critical reading of the manuscript. This work was supported in part by grants from The National Institute of Health (GM61942) to K.Y. and the Dermatology Foundation fellowship to V.A.
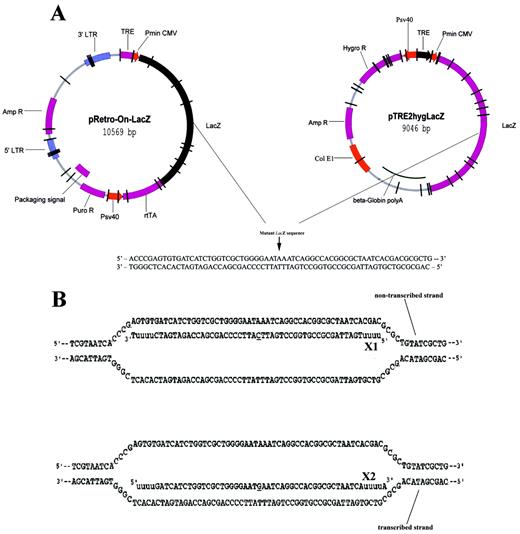
Figure 1. Design of plasmid DNAs and ODNs for episomal targeting in CHO‐K1 and CHO‐AA8‐Luc cells. (A) Diagram of pRetro‐On‐mtLacZ and pTRE2hyg‐mtLacZ plasmids. A point mutation (G→A) at the position 1651 (marked by arrow) causes an amino acid substitution (Glu→Lys), resulting in a complete loss of catalytic activity of the E.coli β‐galactosidase. (B) Hypothetical intermediates between X1 antisense and X2 sense ODNs and the mutant LacZ sequence. Both ODNs were designed to restore an enzymatic activity of the mutant β‐galactosidase by incorporation of a single mismatch to the targeted base (underlined). A double‐stranded target sequence is postulated to be paired with X1 and X2 generating a mismatch A:C and G:T, respectively. The 5′ and 3′ ends of the ODN contain four residues of 2′‐O‐methyl uridine residues (lower case) for protection from nuclease degradation.
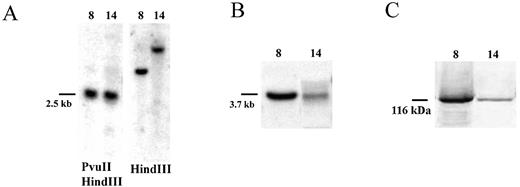
Figure 2. Characterization of two CHO‐K1 clones with integrated mtLacZ gene. (A) Southern blot of the genomic DNA isolated from the CHO‐K1‐mtLacZ clones 8 and 14. (B) Northern blot of RNA isolated from clones 8 and 14. (C) Western blot analysis of the mutant β‐galactosidase protein in clones 8 and 14.
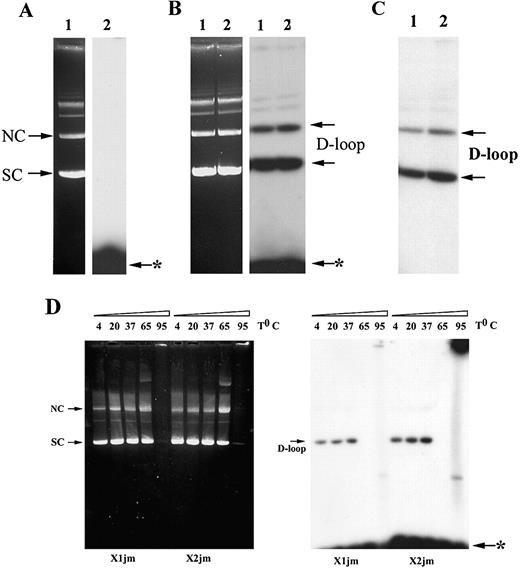
Figure 3. D‐loop formation promoted by RecA. (A) Reaction was performed between pCH110‐G1651A plasmid DNA and the X1 ODN in the absence of RecA. Lane 1 represents ethidium‐bromide staining of the agarose gel and in lane 2 its autoradiogram. (B) Joint formation was performed between pCH110‐G1651A plasmid DNA and the ODN‐RecA pre‐synaptic filament. After deproteinization, synaptic complexes were analyzed on 0.8% agarose gel. The plasmid DNA was visualized by ethidium‐bromide staining (left), whereas joint molecules were detected by autoradiography (right). Lanes 1 and 2 depict the X1‐JM and X2‐JM, respectively. (C) Autoradiogram of the purified joints. Lanes 1 and 2 depict X1‐ and X2‐JM, respectively, after removal of unbound ODN. (D) Stability of D‐loop. After deproteinization, synaptic complexes were incubated at indicated temperatures for 30 min and analyzed on 0.8% agarose gel. Arrows indicate the position of supercoiled DNA (SC), nicked circle (NC) and the D‐loop. Arrow with asterisk indicates unincorporated ODN.
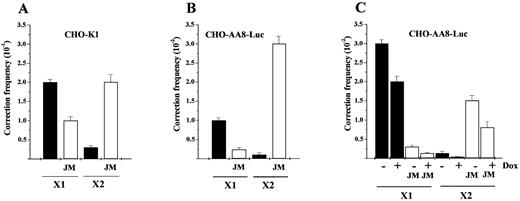
Figure 4. Gene correction of preformed joints in mammalian cells. (A) CHO‐K1 cells were cotransfected with pCH110‐G1651A plasmid and X1 or X2 ODN, respectively. Correction frequency of cotransfection is depicted (closed bar). The corresponding correction frequency of the X1‐JM or X2‐JM is depicted (open bar). (B) CHO‐AA8‐Luc cells were cotransfected with pCH110‐G1651A plasmid and X1 or X2 ODN, respectively. Correction frequency of cotransfection is depicted (closed bar). The corresponding correction frequency of the X1‐JM or X2‐JM is depicted (open bar). (C) CHO‐AA8‐Luc cells were cotransfected with pTRE2hyg‐mtLacZ plasmid and X1 or X2 ODN and transcription was induced by the removal of Dox. Correction frequency of cotransfection is depicted (closed bar). The corresponding correction frequency of the X1‐JM or X2‐JM is depicted (open bar). The frequency was determined from four independent experiments by dividing the number of blue cells containing corrected phenotype by the number of initially targeted cells (5 × 104 cells).
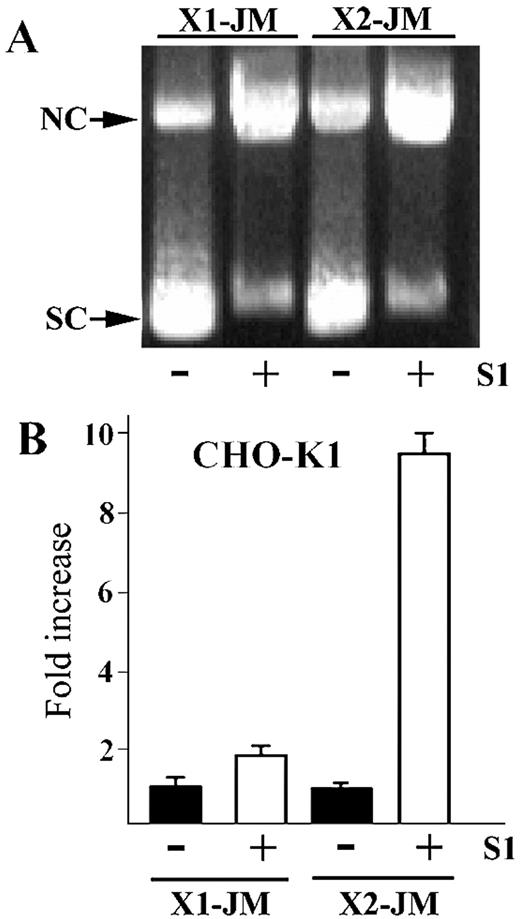
Figure 5. Resected D‐loop and its functional activity in CHO‐K1 cells. (A) The ethidium‐bromide‐stained gel containing the joint (–) and its corresponding resected joint (+) after treatment with S1 nuclease, respectively. Arrows indicate the position of supercoiled DNA (SC) and nicked circle (NC). (B) The resected joint (open bar) shows higher frequency than the joint itself (closed bar). The fold increase was plotted by dividing the number of the X‐gal positive cells in the transfection experiment with the resected joint by those with the joint only.
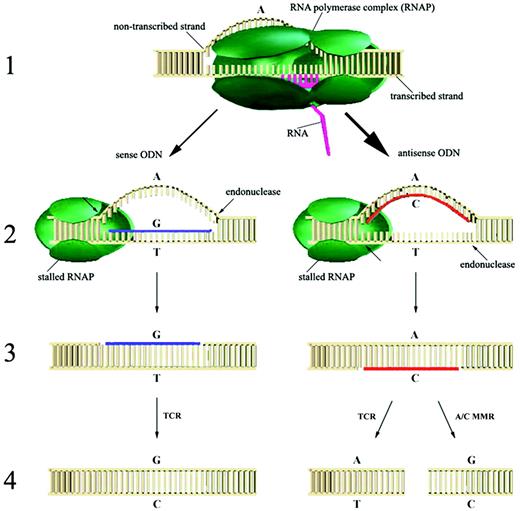
Figure 6. Hypothetical model for the transcription‐coupled ODN‐directed gene repair in mammalian cells (see details in the text).
Episomal gene correction by ODN in the Tet‐On and Tet‐Off systems
| Targeting frequencya | ||
| –Dox | +Dox | |
| Tet‐On ODN | ||
| X1 | (3.4 ± 1.2) × 10–3 | (3.2 ± 1.3) × 10–2 |
| X2 | (2.0 ± 0.9) × 10–3 | (1.9 ± 0.7) × 10–2 |
| Tet‐Off ODN | ||
| X1 | (5.0 ± 1.2) × 10–2 | (2.2 ± 0.6) × 10–2 |
| X2 | (2.6 ± 1.5) × 10–2 | (1.2 ± 0.4) × 10–3 |
| Targeting frequencya | ||
| –Dox | +Dox | |
| Tet‐On ODN | ||
| X1 | (3.4 ± 1.2) × 10–3 | (3.2 ± 1.3) × 10–2 |
| X2 | (2.0 ± 0.9) × 10–3 | (1.9 ± 0.7) × 10–2 |
| Tet‐Off ODN | ||
| X1 | (5.0 ± 1.2) × 10–2 | (2.2 ± 0.6) × 10–2 |
| X2 | (2.6 ± 1.5) × 10–2 | (1.2 ± 0.4) × 10–3 |
aThe frequency of gene correction was determined from five independent experiments by dividing the number of blue cells containing corrected phenotype by the number of initially targeted cells (5 × 104 cells).
Episomal gene correction by ODN in the Tet‐On and Tet‐Off systems
| Targeting frequencya | ||
| –Dox | +Dox | |
| Tet‐On ODN | ||
| X1 | (3.4 ± 1.2) × 10–3 | (3.2 ± 1.3) × 10–2 |
| X2 | (2.0 ± 0.9) × 10–3 | (1.9 ± 0.7) × 10–2 |
| Tet‐Off ODN | ||
| X1 | (5.0 ± 1.2) × 10–2 | (2.2 ± 0.6) × 10–2 |
| X2 | (2.6 ± 1.5) × 10–2 | (1.2 ± 0.4) × 10–3 |
| Targeting frequencya | ||
| –Dox | +Dox | |
| Tet‐On ODN | ||
| X1 | (3.4 ± 1.2) × 10–3 | (3.2 ± 1.3) × 10–2 |
| X2 | (2.0 ± 0.9) × 10–3 | (1.9 ± 0.7) × 10–2 |
| Tet‐Off ODN | ||
| X1 | (5.0 ± 1.2) × 10–2 | (2.2 ± 0.6) × 10–2 |
| X2 | (2.6 ± 1.5) × 10–2 | (1.2 ± 0.4) × 10–3 |
aThe frequency of gene correction was determined from five independent experiments by dividing the number of blue cells containing corrected phenotype by the number of initially targeted cells (5 × 104 cells).
Chromosomal gene correction by ODN in two clones with integrated mtLacZ with different transcriptional levels
| ODN | Targeting frequencya | |
| Clone 8 | Clone 14 | |
| X1 | (1.5 ± 0.3) × 10–3 | (1.5 ± 1.0) × 10–5 |
| X2 | (1.5 ± 0.3) × 10–4 | <0.5 × 10–5 |
| ODN | Targeting frequencya | |
| Clone 8 | Clone 14 | |
| X1 | (1.5 ± 0.3) × 10–3 | (1.5 ± 1.0) × 10–5 |
| X2 | (1.5 ± 0.3) × 10–4 | <0.5 × 10–5 |
aThe frequency of gene correction was determined from five independent experiments by dividing the number of blue cells containing corrected phenotype by the number of initially targeted cells (5 × 104 cells).
Chromosomal gene correction by ODN in two clones with integrated mtLacZ with different transcriptional levels
| ODN | Targeting frequencya | |
| Clone 8 | Clone 14 | |
| X1 | (1.5 ± 0.3) × 10–3 | (1.5 ± 1.0) × 10–5 |
| X2 | (1.5 ± 0.3) × 10–4 | <0.5 × 10–5 |
| ODN | Targeting frequencya | |
| Clone 8 | Clone 14 | |
| X1 | (1.5 ± 0.3) × 10–3 | (1.5 ± 1.0) × 10–5 |
| X2 | (1.5 ± 0.3) × 10–4 | <0.5 × 10–5 |
aThe frequency of gene correction was determined from five independent experiments by dividing the number of blue cells containing corrected phenotype by the number of initially targeted cells (5 × 104 cells).
References
Igoucheva,O. and Yoon,K. (
Rice,M.C., Czymmek,K. and Kmiec,E.B. (
Richardson,P.D., Augustin,L.B., Kren,B.T. and Steer,C.J. (
Igoucheva,O., Peritz,A.E., Levy,D. and Yoon,K. (
Alexeev,V. and Yoon,K. (
Alexeev,V., Igoucheva,O., Domashenko,A., Cotsarelis,G. and Yoon,K. (
Igoucheva,O., Alexeev,V. and Yoon,K. (
Igoucheva,O., Alexeev,V. and Yoon,K. (
Campbell,C.R., Keown,W., Lowe,L., Kirschling,D. and Kucherlapati,R. (
Rauth,S., Song,K.Y., Ayares,D., Wallace,L., Moore,P.D. and Kucherlapati,R. (
Simon,J.R. and Moore,P.D. (
Yamamoto,T., Moerschell,R.P., Wakem,L.P., Komar‐Panicucci,S. and Sherman,F. (
Pierce,E.A., Liu,Q., Igoucheva,O., Omarrudin,R., Ma,H.C., Diamond,S. and Yoon,K. (
Alexeev,V., Igoucheva,O. and Yoon,K. (
Liu,L., Rice,M.C. and Kmiec,E.B. (
Liu,L., Rice,M.C., Drury,M., Cheng,S., Gamper,H. and Kmiec,E.B. (
Chiu,S.K., Low,K.B., Yuan,A. and Radding,C.M. (
Holloman,W.K., Wiegand,R., Hoessli,C. and Radding,C.M. (
Mooney,R.A., Artsimovitch,I. and Landick,R. (
Citterio,E., Vermeulen,W. and Hoeijmakers,J.H. (
Leadon,S.A. (
Tornaletti,S. and Hanawalt,P.C. (
van Gool,A.J., van der Horst,G.T., Citterio,E. and Hoeijmakers,J.H. (
Park,J.S., Marr,M.T. and Roberts,J.W. (
Le Page,F., Kwoh,E.E., Avrutskaya,A., Gentil,A., Leadon,S.A., Sarasin,A. and Cooper,P.K. (
Mellon,I., Rajpal,D.K., Koi,M., Boland,C.R. and Champe,G.N. (
Gupta,R.C., Bazemore,L.R., Golub,E.I. and Radding,C.M. (
Mazin,A.V., Bornarth,C.J., Solinger,J.A., Heyer,W.D. and Kowalczykowski,S.C. (
Van Komen,S., Petukhova,G., Sigurdsson,S., Stratton,S. and Sung,P. (
O’Donovan,A., Davies,A.A., Moggs,J.G., West,S.C. and Wood,R.D. (
Oda,S., Humbert,O., Fiumicino,S., Bignami,M. and Karran,P. (
Modrich,P. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments